当我用DeepSeek预测AI for BioScience未来3-5年发展趋势
预测未来3-5年AI在生物科学(AI for BioScience)的发展趋势,可以从技术突破、跨学科融合、数据驱动创新以及伦理监管等多个维度进行分析。以下是一些关键趋势的展望: 1. 蛋白质与分子设计的革...

一文搞懂易混淆亲缘关系统计量(血缘系数、亲缘系数、近交系数)
背景 血缘系数、亲缘系数、近交系数等表征亲缘关系的统计量,在遗传学中经常使用。然而,这些概念很容易混淆。下面是几个概念的区别。注意的是,这些名词大多由英文翻译而来,而不同地方的中文...
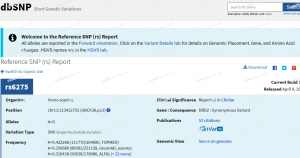
依据SNP染色体和位置信息批量转换rs编号
如果只有SNP的染色体和物理位置信息,该如何批量转换得到 rs ID? 思路非常简单,只需要下载 dbSNP 的参考文件,根据位置信息从参考文件中获取对应的 rs 编号即可。 下面列举两个例子。 重命名 ...
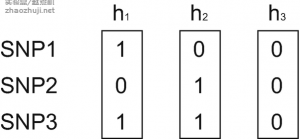
基于RAINBOW的单倍型全基因组关联分析(haplotype-based GWAS)教程
听起来难以置信,单倍型关联分析竟然这么简单 Haplotype-based GWAS(单倍型全基因组关联分析)是基于 haplotype (单倍型)进行的关联分析,在基因组层面寻找与表型相关的变异。 Haplotype 是...
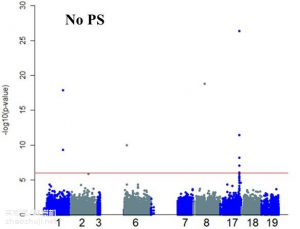
PCA方法校正群体结构,GWAS该用多少个主成分?
该选择多少个主成分 群体结构(population structure),或者说群体分层(population stratification),是由于个体之间非随机交配而导致的群体中亚群之间等位基因频率的系统差异。这种系统差异...

使用ADMIXTURE估计个体的祖先成分
ADMIXTURE 是常用的群体遗传学分析工具,可以估计个体的祖先成分。与 STRUCTURE 相比,它的速度更快。它的下载地址是 http://dalexander.github.io/admixture/download.html,下载解压就可以使...
使用Hapbin基于EHH、iHS、XP-EHH方法检测基因组中的选择信号
EHH(Extended Haplotype Homozygosity)、iHS(Integrated Haplotype Score) 和 XP-EHH(Cross Population Extended Haplotype Homozogysity)是常用的基于 haplotype 分析基因组受选择情况的...
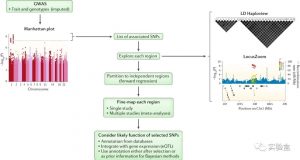
FINEMAP:使用GWAS摘要数据进行无功能注释数据的精细定位(Fine-mapping)
介绍 全基因组关联分析(GWAS)是非常流行的定位表型或疾病遗传位点方法。不过很多情况下,GWAS 发现的最显著的 SNP(top SNP 或者 index SNP)并不是真正造成影响的causal SNP(因果SNP),而...
CAUSALdb:涵盖数千个GWAS研究和Fine-mapping结果的可视化数据库
现在 GWAS 研究越来越多。要查询以往的 GWAS 研究结果,可以使用 GWAS Catalog。GWAS Catalog 包含的信息非常多,不过有时可能满足不了需要。这里,推荐一个叫 CAUSALdb 的数据库。 CAUSALdb(h...

GWAS_Flow:使用GPU加速大规模数据的全基因组关联分析
21世纪是生物的世纪,生物数据的增长速度越来越快。很多分析工具在开发时并没有考虑到大规模数据的应用场景。在数据量不大的时候,这些工具的计算时间并不会太长,可以让人接受。但在数据规模庞...

使用Python版XP-CLR检测基因组中的选择信号
上一篇文章 《使用XP-CLR检测基因组中的选择信号》 介绍了 XP-CLR。XP-CLR 是一种是基于选择扫荡(selective sweeep)的似然方法。选择扫荡可以增加群体之间的遗传分化,导致等位基因频率偏离中...

使用XP-CLR检测基因组中的选择信号
检测基因组选择信号的方法有很多种,其中 XP-CLR 方法是常用的一种。XP-CLR 是陈华老师、Nick Patterson 和 David Reich 在 2010 年发表的方法,全称叫 the cross-population composite likelih...
最近更新