多模态蛋白质语言模型是一个新兴的方向。 《Multi-scale Protein Language Model for Unified Molecular Modeling》是一篇发表在ICML 2024的文献,作者是 Kangjie Zheng、Siyu Long、Tianyu Lu、Junwei Yang、Xinyu Dai、Ming Zhang、Zaiqing Nie、Wei-Ying Ma 和 Hao Zhou。文章的蛋白质语言模型涉及到氨基酸序列和分子结构的多模态信息,提供了一个整合的思路。
摘要
蛋白质语言模型在蛋白质工程领域展现出巨大潜力。然而,现有的蛋白质语言模型主要在残基(氨基酸)尺度上运作,这限制了它们提供原子尺度信息的能力。这种限制阻碍了我们充分利用蛋白质语言模型在涉及蛋白质和小型分子的应用程序中的潜力。因此,作者提出了一种新方法ESM-AA(ESM All-Atom),它通过预训练多尺度代码切换蛋白质序列,并使用多尺度位置编码来捕捉残基和原子之间的关系,从而实现了原子尺度和残基尺度统一的分子建模。实验结果表明,ESM-AA在蛋白质分子任务中超越了先前的方法,展示了蛋白质语言模型的充分利用。进一步的调查揭示了通过统一分子建模,ESM-AA不仅获得了分子知识,还保留了对蛋白质的理解。
引言
蛋白质语言模型在蛋白质工程中展现出巨大潜力,它们在预训练大规模蛋白质序列时能够捕获生化和共同进化知识。这在多个领域取得了显著成就,包括蛋白质结构预测、蛋白质适应度预测和蛋白质设计等。然而,现有的蛋白质语言模型主要在残基尺度上运作,不提供原子尺度的信息。为了充分利用蛋白质语言模型在涉及大分子(蛋白质)和小型分子的应用程序中的潜力,需要将外部小型分子模型纳入考虑。作者提出了ESM-AA,它通过预训练多尺度代码切换蛋白质序列,并使用多尺度位置编码来描述残基和原子之间的关系,实现了多尺度统一分子建模。
方法
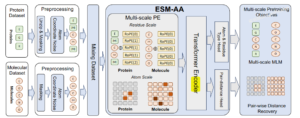
文献提出了提出一个多尺度预训练模型ESM-AA,它通过以下两个主要步骤实现多尺度统一分子建模:1. 在多尺度代码切换蛋白质序列上进行预训练;2. 使用多尺度位置编码描述残基和原子之间的关系。
![图片[1]-ICML 2024|用于统一分子建模的多尺度蛋白质语言模型ESM-AA --实验盒](https://www.shiyanhe.com/wp-content/uploads/2024/05/Pasted-image-20240530090920.png)
模型架构的关键特点和组件如下:
-
多尺度预训练(Multi-scale Pre-training):
- 代码切换蛋白序列(Code-Switch Protein Sequence): 模型通过随机“解压缩”(unzipping)部分残基(氨基酸)到它们的组成原子,并为每个解压缩的原子分配坐标,从而在蛋白质序列中引入原子级别的信息。
- 多尺度位置编码(Multi-scale Position Encoding, MSPE): 为了在代码切换序列中描述残基和原子之间的复杂位置关系,ESM-AA设计了一种多尺度位置编码,包括残基尺度位置编码(Residue Scale Position Encoding, RSPE)和原子尺度位置编码(Atom Scale Position Encoding, ASPE)。
-
残基尺度位置编码(Residue Scale Position Encoding, RSPE):
- 基于现有的编码方法,如Rotary Position Embedding (RoPE),用于描述残基之间的相对位置关系。
- 对于同一残基中的原子,重用该残基的位置编码,避免引入模糊的位置信息。
-
原子尺度位置编码(Atom Scale Position Encoding, ASPE):
- 使用空间距离矩阵和高斯核(Gaussian Kernel)直接对原子的3D位置进行编码,以描述原子之间的关系。
-
变换器编码器(Transformer Encoder):
- ESM-AA采用了标准的Transformer架构,其中位置编码被集成到自注意力层中。
- 自注意力计算时,原子尺度位置编码被视作自注意力层的偏置项(bias term)。
-
预训练任务(Pre-training Objectives):
- 掩码语言建模(Masked Language Modeling, MLM): 模型需要预测被掩盖(masked)的残基或原子。
- 成对距离恢复(Pair-wise Distance Recovery, PDR): 模型需要从被噪声污染的原子坐标中恢复准确的原子间欧几里得距离。
-
多尺度掩码语言建模(Multi-scale MLM):
- 模型同时对残基尺度和原子尺度的标记进行掩码和预测。
-
成对距离恢复(Pair-wise Distance Recovery, PDR):
- 模型需要从被噪声污染的原子坐标中恢复准确的原子间距离。
-
模型参数化(Parameterization):
- ESM-AA使用12层堆叠的Transformer层,每层有20个注意力头。
- 模型维度和前馈维度分别为480和1920。
-
输入处理:
- 模型可以接受蛋白质或分子作为输入,但在预训练中,输入是未配对的蛋白质或分子数据。
-
输出:
- 模型输出可以用于多种下游任务,如蛋白质结构预测、蛋白质适应度预测、蛋白质设计等。
![图片[2]-ICML 2024|用于统一分子建模的多尺度蛋白质语言模型ESM-AA --实验盒](https://www.shiyanhe.com/wp-content/uploads/2024/05/Pasted-image-20240530091035.png)
![图片[3]-ICML 2024|用于统一分子建模的多尺度蛋白质语言模型ESM-AA --实验盒](https://www.shiyanhe.com/wp-content/uploads/2024/05/Pasted-image-20240530091229.png)
ESM-AA模型架构的创新之处在于它能够同时处理蛋白质的残基尺度和原子尺度信息,并通过统一的建模方法提高了蛋白质-分子任务的性能。这种多尺度建模策略使得ESM-AA能够更全面地理解和预测蛋白质及小分子的结构和功能。
实验结果
作者在蛋白质和小型分子的混合数据上预训练ESM-AA,并在多种基准测试上对其进行微调,以验证其性能。主要结果包括:
-
性能提升: ESM-AA在蛋白质分子任务中的表现超越了以往的方法。这表明通过统一的分子建模方法,可以充分利用蛋白质语言模型的潜力。
-
多尺度建模: ESM-AA成功实现了在原子尺度和残基尺度上的统一建模。通过预训练多尺度代码切换蛋白质序列,并使用多尺度位置编码,模型能够同时捕获残基间和原子间的关系。
-
实验验证: 通过一系列实验,作者验证了ESM-AA在多种基准测试上的有效性。这些基准测试包括蛋白质结构预测、蛋白质适应度预测、蛋白质设计等任务。
-
保留蛋白质理解: 尽管ESM-AA被设计为多尺度模型,它仍然保留了对蛋白质的深入理解。这通过在蛋白质特定任务(如二级结构预测和接触预测)上与ESM-2模型相似或更好的性能来证明。
-
分子任务的表现: 当将ESM-AA应用于标准分子基准测试时,它也超越了几个特定于分子的模型,这突出了统一分子建模的潜力。
-
可视化分析: 通过对ESM-AA和ESM-2+Uni-Mol学习到的表示进行可视化比较,结果表明ESM-AA模型能够创建更一致的语义表示,包括蛋白质和分子数据。
-
在虚拟筛选基准测试中的性能: ESM-AA在虚拟筛选基准测试中表现出强大的性能,即使在零样本设置下,也接近最先进的方法DrugCLIP。
-
蛋白质功能注释任务的性能: ESM-AA在蛋白质功能注释任务上的表现与结构蛋白质表示模型相当或更好,即使没有结构信息输入。
-
消融研究: 文献中的消融研究表明,多尺度位置编码、预训练目标和训练数据等各个组成部分对ESM-AA方法的有效性至关重要。
这些结果表明,ESM-AA模型不仅在理论上是创新的,而且在实际应用中也是有效的,能够处理蛋白质和分子的复杂交互,并在多种生物学和化学任务中表现出色。
总结
蛋白质语言模型已广泛应用于多个领域,包括蛋白质结构预测、蛋白质适应度预测和蛋白质设计。统一分子建模扩展了蛋白质语言模型在残基和原子尺度上有效运作的能力,从而增强了它们在这些任务中的适用性。此外,统一分子建模也为蛋白质-小分子相互作用领域的研究开辟了新的途径。基于此,作者提出了ESM-AA,这是一个多尺度蛋白质语言模型,通过预训练多尺度代码切换蛋白质序列和使用多尺度位置编码来描述残基和原子之间的关系,实现了多尺度统一分子建模。实验结果表明,ESM-AA在蛋白质-分子任务中超越了先前的方法,并且有效地将分子知识整合到蛋白质语言模型中,同时没有牺牲对蛋白质的理解。
相关链接
[1] 文献:https://arxiv.org/abs/2403.12995v2
[2] 审稿意见:https://openreview.net/pdf?id=MBIGXMT0qC
![图片[4]-ICML 2024|用于统一分子建模的多尺度蛋白质语言模型ESM-AA --实验盒](https://www.shiyanhe.com/wp-content/uploads/2021/09/wx-banner-website-20210914.png)














暂无评论内容